Data from: The population genomics of archaeological transition in west Iberia: investigation of ancient substructure using imputation and haplotype-based methods Martiniano R, Cassidy LM, Ó'Maoldúin R, McLaughlin R, Silva NM, Manco L, Fidalgo D, Pereira T, Coelho MJ, Serra M, Burger J, Parreira R, Moran E, Valera AC, Porfirio E, Boaventura R, Silva AM, Bradley DG Date Published: July 28, 2017 DOI: http://dx.doi.org/10.5061/dryad.g9f5rKeep in mind however, that this dataset is specifically designed for haplotype-based tests, like those done with Chromopainter (for more details, see S5 Text in Martiniano et al. 2017). As far as I know, it should also perform well in ADMIXTURE runs. On the other hand, the diploid and imputed genotype calls are likely to slightly skew results in formal statistics and formal statistics-based modeling analyses. So it's best to use pseudo-haploid genomes for such tests, and/or high coverage diploid genomes if available, with 100% observed calls. I'm about to run a quick and dirty haplotype/Principal Component analysis with this dataset using BEAGLE, mainly to check whether South Asians show greater recent genetic affinity to Afanasievo/Yamnaya over Andronovo/Sintashta (for more on this controversy, see here). It's a pity that this dataset doesn't include any genomes from Neolithic Iran, because then I'd also be able to try haplotype-based mixture models for South Asians. By the way, I won't be using all of the 30 million markers. I've only kept the SNPs that overlap with the Harvard Medical School's 1240K SNP ancient capture array, which should mean that only a small minority of the calls in my analysis won't be real. Update 02/08/2017: The BEAGLE run is complete and the analysis is unfolding. See post and comments here.
Saturday, July 29, 2017
New resource: 67 diploid ancient genomes
Published this week along with Martiniano et al. 2017, a dataset of 67 new and publicly available genomes, genotyped and imputed for 30 million markers:
Thursday, July 27, 2017
Yamnaya-related migrations into Iberia: infiltration rather than invasion (Martiniano et al. 2017)
The Martiniano et al. preprint that appeared at bioRxiv more than two months ago was published at PLoS Genetics today (see here). The paper packs a lot of supplementary information that wasn't included with the preprint. Below is the press release about the paper from the Public Library of Science (PLoS). Emphasis is mine.
The genomes of individuals who lived on the Iberian Peninsula in the Bronze Age had minor genetic input from Steppe invaders, suggesting that these migrations played a smaller role in the genetic makeup and culture of Iberian people, compared to other parts of Europe. Daniel Bradley and Rui Martiniano of Trinity College Dublin, in Ireland, and Ana Maria Silva of University of Coimbra, Portugal, report these findings July 27, 2017 in PLOS Genetics. Between the Middle Neolithic (4200-3500 BC) and the Middle Bronze Age (1740-1430 BC), Central and Northern Europe received a massive influx of people from the Steppe regions of Eastern Europe and Asia. Archaeological digs in Iberia have uncovered changes in culture and funeral rituals during this time, but no one had looked at the genetic impact of these migrations in this part of Europe. Researchers sequenced the genomes of 14 individuals who lived in Portugal during the Neolithic and Bronze Ages and compared them to other ancient and modern genomes. In contrast with other parts of Europe, they detected only subtle genetic changes between the Portuguese Neolithic and Bronze Age samples resulting from small-scale migration. However, these changes are more pronounced on the paternal lineage. "It was surprising to observe such a striking Y chromosome discontinuity between the Neolithic and the Bronze Age, such as would be consistent with a predominantly male-mediated genetic influx" says first author Rui Martiniano. Researchers also estimated height from the samples, based on relevant DNA sequences, and found that genetic input from Neolithic migrants decreased the height of Europeans, which subsequently increased steadily through later generations. The study finds that migration into the Iberian Peninsula occurred on a much smaller scale compared to the Steppe invasions in Northern, Central and Northwestern Europe, which likely has implications for the spread of language, culture and technology. These findings may provide an explanation for why Iberia harbors a pre-Indo-European language, called Euskera, spoken in the Basque region along the border of Spain and France. It has been suggested that Indo-European spread with migrations through Europe from the Steppe heartland; a model that fits these results. Daniel Bradley says "Unlike further north, a mix of earlier tongues and Indo-European languages persist until the dawn of Iberian history, a pattern that resonates with the real but limited influx of migrants around the Bronze Age."Martiniano R, Cassidy LM, Ó'Maoldúin R, McLaughlin R, Silva NM, Manco L, et al. (2017) The population genomics of archaeological transition in west Iberia: Investigation of ancient substructure using imputation and haplotype-based methods. PLoS Genet 13(7): e1006852. https://doi.org/10.1371/journal.pgen.1006852 See also... New resource: 67 diploid ancient genomes Late PIE ground zero now obvious; location of PIE homeland still uncertain, but...
Thursday, July 20, 2017
The Out-of-India Theory (OIT) challenge: can we hear a viable argument for once?
Recent weeks have seen a rash of activity from OIT proponents defending their "truth", largely as a response to a news feature in The Hindu on new genetic evidence backing the Aryan Invasion or Migration Theory (AIT/AMT). A few examples:
Genetics Might Be Settling The Aryan Migration Debate, But Not How Left-Liberals Believe Genetics and the Aryan invasion debate Propagandizing the Aryan Invasion Debate: A Rebuttal to Tony Joseph Here We Go Again: Why They Are Wrong About The Aryan Migration Debate This Time As Well The problematics of genetics and the Aryan issue Too early to settle the Aryan migration debate?The people who wrote these articles are able to string sentences together in a reasonable way, but apart from that, their efforts are clumsy at best. Not only do they not appear to completely understand what they're attempting to debunk, but they also fail to offer an OIT that realistically incorporates new findings from ancient and modern-day DNA. AIT/AMT is now firmly backed by ancient DNA from Eastern Europe and high resolution modern-day DNA from South Asia. To quote myself from a week ago:
During the past couple of years ancient DNA has revealed the presence of Y-chromosome haplogroup R1a in Eastern European remains dated to the Mesolithic, Neolithic, Eneolithic and Bronze Age. Moreover, the Bronze Age remains, packed in ancestry derived from Eastern European hunter-gatherers (or EHG) and totally lacking any sort of South Asian admixture, belong to R1a-Z645, which is the ancestral clade of by far the most common varieties of R1a in Europe and South Asia today: R1a-Z282 and R1a-Z93, respectively. And on top of that, South Asians, especially those speaking Indo-European languages, show significant admixture derived from EHG. The conclusion from this data is self-evident: during the Bronze Age R1a-Z645 became a very important Y-chromosome lineage in Europe and quickly moved to South Asia, in all likelihood on the back of the Indo-European expansion. Pre-Indo-European Eastern Europe and South Asia were not the same world; they were world's apart. Thus, you will never read anything like this, no matter how much ancient DNA from South Asia is sequenced: During the past couple of years ancient DNA has revealed the presence of Y-chromosome haplogroup R1a in South Asian remains dated to the Mesolithic, Neolithic, Eneolithic and Bronze Age. Moreover, the Bronze Age remains, packed in ancestry derived from South Asian hunter-gatherers, and totally lacking any sort of European admixture, belong to R1a-Z645, which is the ancestral clade of by far the most common varieties of R1a in Europe and South Asia today: R1a-Z282 and R1a-Z93, respectively. And on top of that, Europeans, especially those speaking Indo-European languages, show significant admixture derived from South Asian hunter-gatherers.So, OIT proponents, what counter-arguments can you offer? And can you come up with a new vision for OIT that coherently takes into account ancient DNA from Eastern Europe? However, to ensure that the debate is a fruitful one not derailed regularly by anti-AIT/pro-OIT red herrings, let's take care of the most obvious of these red herrings now. I reserve the right to delete any comments that attempt to go down these tired, irrelevant avenues without a very good excuse for doing so.
You: So and so found Y-haplogroup P* and other basal clades upstream of R1a in Papuans, therefore R1a and Indo-Europeans are from South Asia. Me: Nonsense. R1 and R1a are found in the remains of Eastern European Mesolithic foragers. Were these individuals recently arrived Indo-European-speakers from South Asia? Try harder. You: It doesn't matter that Eastern European Mesolithic foragers belonged to R1a, because the most common form of R1a in the world is R1a-M417, and if it originated in India then OIT is a reality. Me: But what are the chances realistically that R1a-M417 is from India or South Asia, considering that prehistoric European samples, with absolutely no signals of ancestry from South Asia, belong to both M417+ and M417- lineages? In fact, Europe is the most likely homeland of R1a-M417. You: India has incredible diversity in R1a, therefore it's the R1a and Indo-European homeland. Me: No it doesn't. India, and indeed, South Asia as a whole are dominated by one fairly young subclade: R1a-Z93. Europe is home to three different subclades that show up at perceptible frequencies: R1a-Z282, found throughout much of the continent; R1a-L664, mostly confined to Northwestern Europe; and R1a-Z93, mostly confined to far Eastern Europe. You: Many unique Indian ethnic groups are yet to be tested genetically. They may show surprising results, including new subclades of R1a. Me: If you dig hard enough, you'll always find some exceptions to the rule. But how do you know where the ancestral lineages of such exceptions in South Asia were during, say, the Neolithic? What makes you think they were in South Asia? To prove that South Asia is the homeland of its by far most dominant R1a subclade, R1a-Z93, then at the very least you need to show that other, closely and distantly related subclades, are also found at perceptible frequencies in whole regions of South Asia, and therefore that they have some sort of history there. Otherwise we can safely assume that R1a-Z93 and the few exceptions to the R1a-Z93 rule in South Asia are relative latecomers from somewhere else. You: But we have no ancient DNA from South Asia yet, and it may produce a huge shock. Me: For you yes, but not for me. What are the chances realistically that R1a was present among both European and South Asian foragers? I'd say practically zero. Feel free to raise it to a few per cent to make yourself feel better, but we both know the hard reality. You: Ancient DNA from South Asia might show that Northern India was home to a population very similar to Yamnaya, and if so, then the Yamnaya-related ancestry in modern-day Indians is native to India. Me: There's no logic behind this. Yamnaya and other closely related Bronze Age groups were very specific mixtures of Mesolithic foragers and Neolithic farmers living in Eastern Europe and surrounds. There's absolutely no reason to assume that such unique mixtures would also form independently in South Asia, or even outside of Europe's generally accepted borders. You: Bronze Age Europeans who belonged to R1a also carried southern admixture from Iran, or maybe even India. Me: In prehistoric samples, R1a is always highly correlated with Eastern European Hunter-Gatherer (EHG) ancestry, so positing that it also arrived in Europe with a southern population makes no sense. And why would this southern ancestry be from Iran or India? Why not the Caucasus? We know from ancient DNA that the type of southern ancestry that these ancient Europeans carried has been sitting in the Caucasus since the Upper Paleolithic. Moreover, they lack South Caspian- and South Asian-specific markers such as mtDNA haplogroup U7. How were such markers purged from their gene pool if they or their recent ancestors arrived in Europe from Iran or India? You: Chickens and mice came from South Asia, therefore Indo-Europeans came from South Asia too. Me: Bullshit. Do better or go away.Does anyone want to claim that I don't know what I'm talking about? Or perhaps that I'm just putting out Eurocentric propaganda? If you don't understand my arguments, and that they're indeed very solid arguments, then there's no hope for you. Go and find a new hobby or profession, because you're not cut out for this. OK, now what we have the formalities out of the way, who wants to have a go at salvaging OIT in the comments? Don't be shy. See also... Ancient herders from the Pontic-Caspian steppe crashed into India: no ifs or buts R1a-M417 from Eneolithic Ukraine!!!11 Late PIE ground zero now obvious; location of PIE homeland still uncertain, but...
Monday, July 17, 2017
On the Mesolithic colonization of Scandinavia (Günther et al. 2017 preprint)
Over at bioRxiv at this LINK. The main takeaway point from this preprint is that Scandinavia was a more happening place than most of the rest of Europe during the Mesolithic, because at the time it was the meeting place between two relatively divergent forager groups, West European hunter-gatherers (WHG) and East European hunter-gatherers (EHG), that entered the peninsula from different directions, the southwest and northeast, respectively, and mixed to form Scandinavian hunter-gatherers (SHG). Other key points:
- EHG probably dispersed across Scandinavia in a counter-clockwise direction via an ice-free route along the Atlantic coast in what is now Norway, because SHG samples from northern and western Scandinavia show more EHG ancestry than those from southern and eastern Scandinavia - at least 17% of the SNPs that are common in SHG are not found in present-day Europeans, suggesting that a large part of European variation has been lost since the Mesolithic - although it's unlikely that SHG made a significant contribution to the present-day Northern European gene pool, some gene-variants common in SHG that appear to be associated with metabolic, cardiovascular, developmental and psychological traits are carried at high frequencies by present-day Northern Europeans, especially compared to present-day Southern Europeans, probably due to strong selective pressures specific to northern latitudes in Europe - SHG is inferred to have had fair skin and varied blue to light-brown eye color, which makes sense considering that it was a mixture of apparently fair-skinned/brown-eyed EHG and dark-skinned/blue-eyed WHG, except that the frequencies of blue-eyed variants and one fair-skinned variant in SHG are much higher than expected from its EHG/WHG mixture ratios, again pointing to strong selective pressures specific to northern latitudes in Europe acting upon certain gene-variants - a 3D computer generated facial reconstruction of an SHG female based on data from a very high (57x) coverage genome sequence looks, at least to me, like a fairly typical present-day Northern European woman (see Figure S9.1 in the supp info here), though I suspect that the result might be biased in some way, simply because it's impossible to know whether variants associated with specific facial traits in present-day Northern Europeans were also associated with the same facial traits in SHG.Citation... Günther et al., Genomics of Mesolithic Scandinavia reveal colonization routes and high-latitude adaptation, bioRxiv, Posted July 17, 2017, doi: https://doi.org/10.1101/164400
Sunday, July 16, 2017
North European admixture in the Han Chinese (Charleston et al. 2017 preprint)
Over at bioRxiv at this LINK. Emphasis is mine. The estimated date of the North European-related admixture signal is probably much too late. These sorts of estimates always look way off. And I doubt that it's largely the result of the Silk Road, which linked China to the Near East and Mediterranean rather than to Northern Europe. More likely it reflects gene flow from the Pontic-Caspian steppe in Eastern Europe during the Bronze and Iron ages, via the Afanasievo, Andronovo, and other closely related steppe peoples (see here).
Abstract: As are most non-European populations around the globe, the Han Chinese are relatively understudied in population and medical genetics studies. From low-coverage whole-genome sequencing of 11,670 Han Chinese women we present a catalog of 25,057,223 variants, including 548,401 novel variants that are seen at least 10 times in our dataset. Individuals from our study come from 19 out of 22 provinces across China, allowing us to study population structure, genetic ancestry, and local adaptation in Han Chinese. We identify previously unrecognized population structure along the East-West axis of China and report unique signals of admixture across geographical space, such as European influences among the Northwestern provinces of China. Finally, we identified a number of highly differentiated loci, indicative of local adaptation in the Han Chinese. In particular, we detected extreme differentiation among the Han Chinese at MTHFR, ADH7, and FADS loci, suggesting that these loci may not be specifically selected in Tibetan and Inuit populations as previously suggested. On the other hand, we find that Neandertal ancestry does not vary significantly across the provinces, consistent with admixture prior to the dispersal of modern Han Chinese. Furthermore, contrary to a previous report, Neandertal ancestry does not explain a significant amount of heritability in depression. Our findings provide the largest genetic data set so far made available for Han Chinese and provide insights into the history and population structure of the world's largest ethnic group. ... One finding from our analysis of admixture signals that most likely fit a one-pulse admixture model is our observation of admixture from Northern European populations to the Northwestern provinces of China (Gansu, Shaanxi, Shanxi), but not other parts of China. Previous analysis of the HGDP data, based on patterns of haplotype sharing among 10 Han Chinese from Northern China, estimated a single pulse of ~6% West Eurasian ancestry among the Northern Han Chinese. The estimated date of admixture was around 1200 CE. This signal is also observed among the Tu people, an ethnic minority also from Northwestern China; the authors attributed this signal to contact through the Silk Road (Hellenthal et al. 2014). We estimate a lower bound of admixture proportion due to Northern Europeans at approximately 2%-5%, with an admixture date of about 26 +/-3 generations for Gansu, and 47 +/-3 generations for Shaanxi [Table S8]. Using a generation time of about 26-30 years (Moorjani et al. 2016), these estimates correspond to admixture events occurring at around 700 CE and 1300 CE, respectively, corresponding roughly to the Tang and Yuan dynasty in China. However, these estimated dates should be interpreted with caution, as both the violation of a single pulse admixture model and the additional noise in inter-marker LD estimates due to low coverage data could bias the estimates.Charleston et al.,A comprehensive map of genetic variation in the world's largest ethnic group - Han Chinese, bioRxiv, Posted July 13, 2017, doi: https://doi.org/10.1101/162982 See also... Late PIE ground zero now obvious; location of PIE homeland still uncertain, but...
Wednesday, July 12, 2017
Indian confirmation bias
In a largely fact free but obfuscation rich comment piece at The Hindu, Indian scientists Gyaneshwer Chaubey and Kumarasamy Thangaraj ask whether it's too early to settle the Aryan migration debate. See here.
No, it's not too early. It's game over chaps, and has been for a while.
During the past couple of years ancient DNA has revealed the presence of Y-chromosome haplogroup R1a in Eastern European remains dated to the Mesolithic, Neolithic, Eneolithic and Bronze Age. Moreover, the Bronze Age remains, packed in ancestry derived from Eastern European hunter-gatherers (or EHG) and totally lacking any sort of South Asian admixture, belong to R1a-Z645, which is the ancestral clade of by far the most common varieties of R1a in Europe and South Asia today: R1a-Z282 and R1a-Z93, respectively. And on top of that, South Asians, especially those speaking Indo-European languages, show significant admixture derived from EHG.
The conclusion from this data is self-evident: during the Bronze Age R1a-Z645 became a very important Y-chromosome lineage in Europe and quickly moved to South Asia, in all likelihood on the back of the Indo-European expansion. Yet, in spite of this, Gyaneshwer and Kumarasamy make the following claim in their article.
Moreover, there is evidence which is consistent with the early presence of several R1a branches in India (our unpublished data).Potentially powerful stuff, you might say. But hang on, what are Gyaneshwer and Kumarasamy seeing in their data that could possibly reverse the current reality about R1a? Did they find R1a in South Asian remains from the Mesolithic and Neolithic? Or perhaps they've uncovered South Asian Bronze Age remains that belong to R1a-Z645 and lack any signals of ancestry from Eastern Europe? This is impossible. The ancient DNA from Eastern Europe says so. That's because pre-Indo-European Eastern Europe and South Asia were not the same world; they were world's apart. Thus, you will never read anything like this, no matter how much ancient DNA from South Asia is sequenced:
During the past couple of years ancient DNA has revealed the presence of Y-chromosome haplogroup R1a in South Asian remains dated to the Mesolithic, Neolithic, Eneolithic and Bronze Age. Moreover, the Bronze Age remains, packed in ancestry derived from South Asian hunter-gatherers, and totally lacking any sort of European admixture, belong to R1a-Z645, which is the ancestral clade of by far the most common types of R1a in Europe and South Asia today: R1a-Z282 and R1a-Z93, respectively. And on top of that, Europeans, especially those speaking Indo-European languages, show significant admixture derived from South Asian hunter-gatherers.See also... The resistance crumbles Ancient herders from the Pontic-Caspian steppe crashed into India: no ifs or buts The Out-of-India Theory (OIT) challenge: can we hear a viable argument for once?
Tuesday, July 11, 2017
Working topology for Eurasian population structure
Here's my new "basic" qpGraph topology that I'll be using to test phylogenetic and mixture models for Eurasians. I think it reconciles a few key findings from recent scientific literature. Please note that since my main interest is post-Neolithic prehistory of West Eurasia, and in particular the early Indo-European expansions, I don't want to make this model unnecessarily complex by adding "dead end" Upper Paleolithic genomes.
 But I welcome ideas on how to improve and make use of this topology, so if, say, adding Ust_Ishim helps, then let's do it. The ancient samples featured in the above graph are listed here and the graph file is available here. Feel free to post your own versions of the graph file in the comments and I'll run them as soon as possible. But please remember to label the samples correctly at all times.
Update 13/07/2017: Thanks to Matt in the comments, here's a neater version of the same model, with a lower (highest) Z score and slightly different mixture coefficients. It includes a couple of zero edges, which are generally undesirable, but these might disappear when more populations are added to the topology. The graph file is available here.
But I welcome ideas on how to improve and make use of this topology, so if, say, adding Ust_Ishim helps, then let's do it. The ancient samples featured in the above graph are listed here and the graph file is available here. Feel free to post your own versions of the graph file in the comments and I'll run them as soon as possible. But please remember to label the samples correctly at all times.
Update 13/07/2017: Thanks to Matt in the comments, here's a neater version of the same model, with a lower (highest) Z score and slightly different mixture coefficients. It includes a couple of zero edges, which are generally undesirable, but these might disappear when more populations are added to the topology. The graph file is available here.



Monday, July 10, 2017
Armenian confirmation bias
Current Biology recently published a paper by Margaryan and Derenko et al. titled Eight Millennia of Matrilineal Genetic Continuity in the South Caucasus. I wasn't going to bother calling out the authors on their, unfortunately I have to say, rather dubious claim, but then I saw this ScienceNordic article enthusiastically attempting to drive home their misguided point, so a few words are now in order.
“It’s basically the same female population in the region over the past 8,000 years. It’s very surprising considering the many waves of migration and cultural shifts,” says lead-author Ashot Margaryan from the Centre for GeoGenetics at the National History Museum of Denmark, University of Copenhagen. Genetics have remained constant for 8,000 years in world’s melting potI'm at a loss as to why Ashot Margaryan is very surprised. I'm not even mildly surprised. Why? Let's take a closer look at what we're dealing with here:
- the authors sequenced just 52 full mitogenomes to represent 8,000 years of prehistory and early history in the South Caucasus - they lumped all of these sequences together into an "Ancient" sample set as if they were from a single time slice (I know, pretty crazy) - they then ran a few complex models on this neither here nor there sample set, and concluded that it resembled the maternal gene pool of present-day Armenians.Well, duh, present-day Armenians are more or less the end product of the population history of the last eight thousand years in what is now Armenia and surrounds. Is anyone still as surprised about this as Ashot? Surely not. Obviously, the problem here is that the authors have mistaken their none too surprising outcome to mean that the South Caucasus has not experienced any major upheavals in its maternal gene pool over the past 8,000 years, which, if actually true, would indeed be very surprising, and even shocking. But the haplogroup assignments of the 52 mitogenomes are reported in the spreadsheet here, and just by eyeballing these results, I can tell that they suggest an influx of foreign ancestry, probably from the Pontic-Caspian steppe, to the South Caucasus after the Early Bronze Age (EBA). Note, for instance, the presence of what appear to be typically steppe haplogroups U4a, U2e1e and U5a1b in the samples dated to the Middle Bronze Age (MBA), Late Bronze Age (LBA) and Early Iron Age (EIA), respectively. Citation... Margaryan and Derenko et al., Eight Millennia of Matrilineal Genetic Continuity in the South Caucasus, Current Biology 27, 1–6 July 10, 2017, DOI: 10.1016/j.cub.2017.05.087
Tuesday, July 4, 2017
Out-of-India chickens coming home to roost
Razib has posted a spacious but none-too-technical review of the ongoing Aryan Invasion Theory (AIT) controversy, along with some personal anecdotes and predictions about how ancient DNA from South Asia might shape the debate in the near future (see here).
It should be a useful guide to the topic for those of you who aren't quite as excited about reading about my latest adventures with qpGraph as many of the regulars in the comments here.
One thing that I'd perhaps add to Razib's post is that the ancient DNA record now boasts Late Neolithic Yamnaya-like Corded Ware Culture individuals from the East Baltic region that belong to Y-haplogroup R1a-Z645. And that's usually as far as their lineages go (see here).
This is important, because the Z645 mutation is directly and recently ancestral to the pair of likely post-Neolithic mutations that define the two R1a subclades most common in Europe and South Asia today: Z282 and Z93, respectively.
So not only are the "European" R1a-Z282 and "South Asian" R1a-Z93 relatively young sister clades, but their ancestral clade has now been found in ancient samples from Northeastern Europe that probably predate their appearance by only a few generations, if that. Of course, the upshot of all of this is that R1a-Z93 could not have originated very far from the East Baltic, which makes South Asia look about as likely as the homeland of this subclade as the goddamn moon. Conversely, it makes AIT look very plausible indeed.
However, granted, this might seem very confusing to anyone who hasn't been studying the R1a topology for years, and perhaps better left out of the more mainstream debates on AIT for the sake of simplicity. By the way, I found this part of Razib's post especially intriguing:
One scientist who holds to the position that most South Asian ancestry dates to the Pleistocene argued to me that we don’t know if ancient Indian samples from the northwest won’t share even more ancestry than the Iranian Neolithic and Pontic steppe samples. In other words, ANI was part of some genetic continuum that extended to the west and north. This is possible, but I do not find it plausible.I suspect that this scientist's rather fanciful suggestion (which really flies in the face of very solid models based on ancient genomic data from Europe and surrounds) is a hint of the direction that the debate will take right after the publication of ancient genomes from South Asia. Because when that happens, obfuscators like this guy (usually hopeless Out-of-India proponents) will either have to concede defeat and quit the debate, or ramp up their obfuscations to spectacular new highs. And please don't mistake my confidence on this issue for bluff and bluster. It's not exactly the best kept secret out there that ancient samples from India and Pakistan are now ready, and...oops I probably can't say more than that for now. Pity. See also... Ancient herders from the Pontic-Caspian steppe crashed into India: no ifs or buts Indian confirmation bias
Europeans: genetically homogeneous on a global scale
From SMBE 2017 via benmpeter on Twitter:
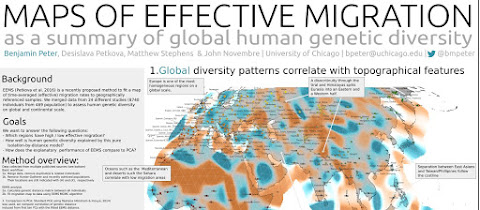 Also at SMBE 2017, David Reich is "sad to leave space of f-statistics", presumably because they don't offer enough resolution when analyzing more recent ancient data from such genetically homogeneous regions as Europe. Via jgschraiber on Twitter.
Update 04/07/2017: A PDF of the Benjamin Peter poster is available at figshare here (30MB).
Also at SMBE 2017, David Reich is "sad to leave space of f-statistics", presumably because they don't offer enough resolution when analyzing more recent ancient data from such genetically homogeneous regions as Europe. Via jgschraiber on Twitter.
Update 04/07/2017: A PDF of the Benjamin Peter poster is available at figshare here (30MB).
 See also...
SMBE 2017 abstracts
Matters of geography
See also...
SMBE 2017 abstracts
Matters of geography


Monday, July 3, 2017
The Indo-Europeanization of South Asia: migration or invasion?
The recent avalanche of ancient DNA data from across Eastern Europe, including modern-day Bulgaria, Estonia, Latvia, Romania, Ukraine and western Russia, has revealed prehistoric hunter-gatherer populations indigenous to the region harboring a remarkable diversity in Y-chromosome lineages belonging to haplogroups R1, R1a and R1b.
Neolithic transition in the Baltic Baltic Corded Ware: rich in R1a-Z645 The genetic history of Northern Europe The genomic history of Southeastern Europe A few more ancient genomes from the Balkans and IberiaSo the once popular idea that these Y-haplogroups were instead native to Central Asia, the Near East and/or South Asia now looks very wrong. R1a probably first arrived in South Asia during the Bronze Age with highly mobile Yamnaya-related pastoralists. These people were expanding in almost all directions from the Pontic-Caspian steppe at the time, and it's difficult to imagine that they weren't the ones who first spread Indo-European languages to peninsular Europe and the Indian subcontinent. It's likely that almost all interested parties will soon agree that this was indeed the case. So the focus in the debate on the expansion of the Indo-Europeans, including Indo-Aryans, into South Asia will soon have to shift from whether it actually happened to how it happened. For instance, was it simply a migration or potentially violent invasion? I already strongly believe that it was an invasion, or rather a series of invasions. I'll change my mind if, at the end of the day, the evidence says otherwise. But if you favor a migration scenario, then consider these points:
- the population in the northern part of the Indian subcontinent during the Bronze Age, even after the collapse of the Indus Civilization, was likely to have been very large for its time, and yet there was a massive pulse of admixture across South Asia from the steppe and a turnover in Y-chromosomes, especially amongst the ruling classes, suggesting that something very dramatic took place that had a major impact on the social and political fabric of the region - early Indo-Europeans in the Near East, from the Hittites to the Scythians, are often recorded as warlike and expansionist, with a habit of invading and subjugating other peoples, like the Hattians, Hurrians and Mitanni (who apparently ended up with an Aryan elite) - if early Indo-Europeans outside of South Asia had a penchant for invasions, then there's no reason to believe that the M.O. of the early Indo-Europeans in South Asia would have been any different, unless some sort of direct empirical evidence says so, but what kind of direct empirical evidence?Please note, I agree that the suggestion of a potentially violent invasion of South Asia by Indo-Europeans, and, indeed, Aryans, sounds provocative, and will always be politically controversial no matter how much evidence is gathered in its favor. But what if it really happened? See also... Ancient herders from the Pontic-Caspian steppe crashed into India: no ifs or buts Indian confirmation bias
Subscribe to:
Comments (Atom)